Скачать презентацию
Идет загрузка презентации. Пожалуйста, подождите

2
Prepared by: Leila Mohammadi 6 th course Supervisors: Prof. E.A.Kogan Prof. N.I.Bubionova

3
DEFINITION Rhabdomyoma is a lesion of striated muscle. The 2 types of rhabdomyoma are neoplastic and hamartoma. The neoplastic variety is subclassified into adult, fetal, and genital types. Hamartomas are divided into cardiac rhabdomyoma and rhabdomyomatous mesenchymal hamartomas of the skin. Cardiac tumor occur very rarely in infancy. When present, the commonest of these are rhabdomyoma. Until now, only a few cases of rhabdomyoma have been diagnosed echocardiographically. Affected infants with severe involvement usually die as a result of obstruction to blood flow, and thus operation may be undertaken in an attempt at palliation. Tuberous sclerosis is often associated with rhabdomyoma and must be sought.

4
Cardiac rhabdomyoma is the most common benign congenital cardiac tumor. It is thought that the tumor is a hamartoma rather than a true neoplasm This is the most common tumor in infants and children. Cardiac rhabdomyoma is caused by mutations in the TSC1 and TSC2 genes. The tumor is closely associated with tuberous sclerosis and occurs in more than 50% of patients with this disease. Such complications occur almost exclusively during pregnancy or within the child's first year. Prenatal ultrasound, performed by an obstetric sonographer specializing in cardiology, can detect a rhabdomyoma after 20 weeks. This rare tumor is a strong indicator of TSC in the child, especially if there is a family history of TSC. Site: Any chamber of the heart may be affected. The left ventricle is the most frequently involved site.

5
Tuberous sclerosis or tuberous sclerosis complex (TSC) is a rare, multi-system genetic disease that causes benign tumors to grow in the brain and on other vital organs such as the kidneys, heart, eyes, lungs, and skin. A combination of symptoms may include seizures, developmental delay, behavioral problems, skin abnormalities, lung and kidney disease. TSC is caused by mutations on either of two genes, TSC1 and TSC2, which encode for the proteins hamartin and tuberin respectively. These proteins act as tumor growth suppressors, agents that regulate cell proliferation and differentiation.
 is a rare, multi-system genetic disease that causes benign tumors to grow in the brain and on other vital organs such as the kidneys, heart, eyes, lungs, and skin. A combination of symptoms may i")
6
Facial angiofibroma, previously termed adenoma sebaceum, in a patient with tuberous sclerosis complex (TSC).
.")
7
MRI of a patient with tuberous sclerosis complex (TSC) demonstrates the presence of a tuber and subependymal nodules.
 demonstrates the presence of a tuber and subependymal nodules.")
8
HISTORY The name of the disease, composed of the Latin word tuber (swelling, nodule) and the Greek word skleros (hard), refers to the pathological finding of thick, firm and pale gyri, called "tubers", in the brains of patients postmortem. These tubers were first described by Désiré-Magloire Bourneville in 1880; the cortical manifestations may sometimes still be known by the eponym Bourneville's disease.
 and the Greek word skleros (hard), refers to the pathological finding of thick, firm and pale gyri, called")
9
EPIDEMIOLOGY Cardiac rhabdomyoma is observed in men and women equally. Primary paediatric cardiac tumors are rare, with an estimated incidence of 0.27 % at autopsy with the most common being, rhabdomyoma. Prevalence rate of tuberous sclerosis is 0.3 to 1 case per newborns.

10
Hamartin and tuberin function as a complex which is involved in the control of cell growth and cell division. (The complex appears to be a Rheb GTPase which suppresses mTOR signaling, part of the growth factor (insulin) signaling pathway.) Thus, mutations at the TSC1 and TSC2 loci result in a loss of control of cell growth and cell division, and therefore a predisposition to forming tumors.
 signaling pathway.) Thus, muta")
11
Many studies in animals demonstrated that Tuberous Sclerosis C2+/- loss or inhibition is associated with up-regulation of a peptide named cyclin D1, regulated by a beta-catenin and the results were that the cell proliferation effects of hamartin and tuberin are mediated through beta-catenin protein and the high levels of beta-catenin were linked to renal tumors as angiomyolipoms in Tuberous Sclerosis C2 mutations. So the hypothesis is that hamartin and tuberin regulates negatively the beta-catenin stability and activity by the deregulation of the beta- catenin complex. The localization and the distribution of hamartin and tuberin are different in different tissues. In general, hamartin and tuberin are expressed in epithelium cells, myocytes and neurons. Besides seems that the co-expressing hamartin and tuberin tissues are prone to a higher incidence of hamartomas than those expressing only one protein but in different patterns of cellular localization.

12
Cyclin D1 TSC2 B- catenin Note: B-catenin Renal angiomyolipoma Almost 50% of TSC patients suffer from renal angiomyolipoma.

14
GENETICS Tuberous sclerosis is inherited in an autosomal dominant fashion.

15
TSC1 encodes for the protein hamartin, is located on chromosome 9 q34. TSC2 encodes for the protein tuberin, is located on chromosome 16 p13.3. TSC1 and TSC2 are both tumor suppressor genes that function according to Knudson's "two hit" hypothesis. That is, a second random mutation must occur before a tumor can develop. This explains why, despite its 100 percent penetrance, TSC has wide expressivity. KNUDSONS HYPOTHESIS: Cancerogenesis depends on: 1. Activation of proto-oncogenes genes 2. Deactivation of tumor suppressor genes

16
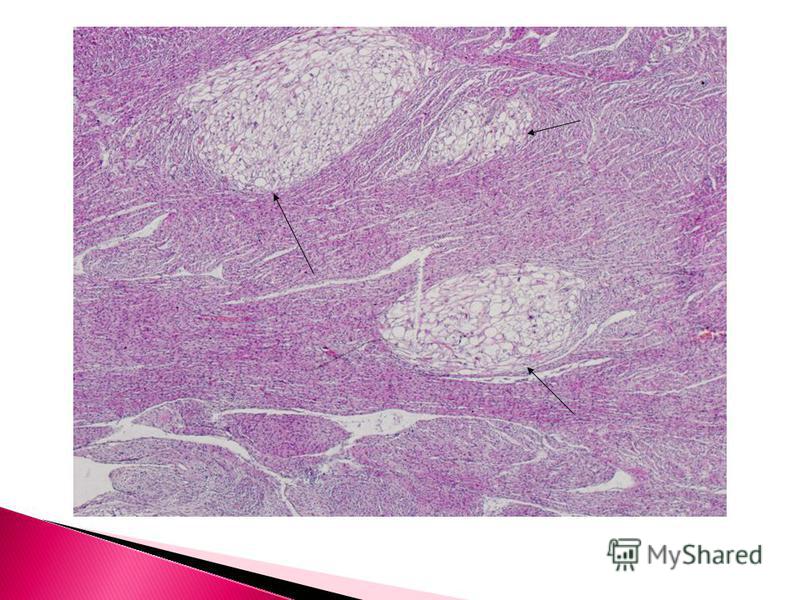
GROSS FEATURES The tumor may present as a single or multiple, non-capsulated soft lesion which can be easily distinguished from the surrounding myocardium. Diffuse rhabdomyoma is extremely rare. Large tumors may show intracavitary extension with almost obliteration of the cavity (D/D: may be confused with cardiac myxoma). Microscopic method.Cells stain strongly with periodic-acid Schiff stains due to their high glycogen content Microscopic features Histologically the tumor is composed of spider cells'. Spider cells are large swollen myocytes with clear cytoplasm and centrally placed cytoplasmic mass and nucleus, connected with the periphery of the cell by strands of cytoplasm. The spider cells are surrounded by normal cardiac myocytes.

18
FATE OF THE TUMOR Cardiac rhabdomyoma decreases in size with age. Echocardiographic evaluation has confirmed the spontaneous regression with eventual disappearance of cardiac rhabdomyoma. Hence, cardiac surgery should be delayed to see whether or not the tumour would regress. Such biologic behavior supports the concept that cardiac rhabdomyomas are hamartoma rather than a true neoplasm.

19
CLINICAL PRESENTATION Clinical profile varies from still-birth to intrauterine myocardial infarct (due to coronary artery compression). Cardiac rhabdomyoma is frequently multiple and asymptomatic, but the tumor may cause cardiomegaly, heart failure by causing outflow obstruction, arrhythmias, thromboembolic disease or sudden unexpected death. Patients with cardiac rhabdomyoma may present with a history of shortness of breath, sometimes associated with signs and symptoms suggestive of cerebral palsy (suggesting the possibility of associated tuberous sclerosis). Patients with cardiac rhabdomyomas may present with heart murmurs. If tuberous sclerosis is associated, the patient displays cerebral palsy–type signs. Renal functions may be altered.
. Cardiac rhabdomyoma is frequently multiple and asymptomatic, but the tumor may cause cardiomegaly, heart failure b")
20
Transabdominal Ultrasound (from 2 nd trimester) Echocardiography (infancy) Computing Tomography (CT) and MRI of the heart are the main non-invasive diagnostic tools. Open surgical or endomyocardial biopsy is only utilized to reveal the histology of the lesion before surgical resection.
 Echocardiography (infancy) Computing Tomography (CT) and MRI of the heart are the main non-invasive diagnostic tools. Open surgical or endomyocardial biopsy is only utilized to reveal the histology of t")
21
DIFFERENTIAL DIAGNOSIS myxoma, hemangioma, Teratoma, Purkinje cell tumor (Foamy myocardial transformation) mesothelioma Glycogen storage disease (shape of the cardiomyocytes are well preserved ).
 mesothelioma Glycogen storage disease (shape of the cardiomyocytes are well preserved ).")
22
PROGNOSIS The tumor(s) appear to exhibit a biphasic growth. They grow until about 32 weeks of gestation and then shrinks progressively during the first year of life. The period of rapid intrauterine growth has been associated with high gestational hormone concentrations, which might cause hyperplastic and hypertrophic response in the tumors. Spontaneous regression has been reported both in utero and childhood. Sudden death is not uncommon % of infants die before reaching their second year. Prognosis ultimately depends on a variety factors including size, associated arrhythmias, and the exact location of the tumor. Inflow or outflow obstruction may lead to congestive cardiac failure, atrioventricular valve dysfunction with valvular incompetence.
 appear to exhibit a biphasic growth. They grow until about 32 weeks of gestation and then shrinks progressively during the first year of life. The period of rapid intrauterine growth has been associated with high gestational ho")
23
Patient is a 29 y.o lady which is G3 and P2, L2 Her past medical history is as follow: varicose in the leg Papilomatous vulvitis ARVI- in 16 th week of pregnancy She had an uncomplicated pregnancy and she has had all routine tests and sonographies. In 26 th week of her pregnancy in the trans abdominal ultrasound some defects of the fetus were detected which are as follows: Agenesis of corpus collosum Hypotrophy of 1 st step Cardiac malformation

24
Based on sonographic data which was done on 26 th week about the malformations the termination of pregnancy was done in 27 th week. PATHOLOGICO-ANATOMICAL DIAGNOSIS: Agenesis of corpus collusum, multiple cardiac rhabdomyomas. Antenatal fetus asphyxia, venous congestion and parenchymal dystrophy of the internal organs, points of hematoma on peritoneum, pleura and pericardium. Total lung athelectasis.

25
MACROSCOPIC FEATURES: F, 984 gr, 34 cm. Cyanotic skin and mucous membrane appearance. Poor development of muscles. corpus collusum is absent. Heart - in LT ventricle, in papillary muscle and mitral valve some nodules with elastic consistence 0.1 to 0.6 cm in diameter were detected

26
MICROSCOPIC FEATURES: Heart: immature dystrophic changes in cardiomyocytes, multiple nodules which contain oval and round cells with small bright nucleus and have glycogenic cytoplasm. Lung: immature, large athelectasis, venous congestion Kidney: immature (2-3 columns of glumeruli ), full of blooded vessels. Liver: dystrophy of hepatocytes, extra medullar lesions. Brain: dystrophic changes in neurons, pericellular edema

27
Tuberous sclerosis complex with involvement of the following organs: Cardiac rhabdomyoma Brian Kidney Liver

31
^ Bader RS, Chitayat D, Kelly E, et al (November 2003). "Fetal rhabdomyoma: prenatal diagnosis, clinical outcome, and incidence of associated tuberous sclerosis complex". J. Pediatr. 143 (5): 620–4. PMID ^ Pérez-Alonso P, Sánchez-Simón R, Contreras F, Patrón-Romero M (December 2000). "Special feature: pathological case of the month. Denouement and discussion: fetal rhabdomyoma of the tongue (myxoid type)". Arch Pediatr Adolesc Med 154 (12): 1265–6. PMID ^ Sugiyama H, Naito H, Tsukano S, Echigo S, Kamiya T (November 2005). "Evaluation of cardiac tumors in children by electron-beam computed tomography: rhabdomyoma and fibroma". Circ. J. 69 (11): 1352–6. PMID ^ European Chromosome 16 Tuberous Sclerosis Consortium (1993). "Identification and characterization of the tuberous sclerosis gene on chromosome 16". Cell 75 (7): 1305–15. PMID ^ Brook-Carter PT, et al (1994). "Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease--a contiguous gene syndrome". Nature Genetics 8 (4): 328–32. doi: /ng PMID ^ Rendtorff ND, et al (2005). "Analysis of 65 tuberous sclerosis complex (TSC) patients by TSC2 DGGE, TSC1/TSC2 MLPA, and TSC1 long-range PCR sequencing, and report of 28 novel mutations". Human Mutation 26 (4): 374–83. doi: /humu PMID ^ a b Roach E, Sparagana S (2004). "Diagnosis of tuberous sclerosis complex". Journal of Child Neurology 19 (9): 643–9. PMID Sites: emedicine.medscape.com/article/ overview en.wikipedia.org/wiki/Rhabdomyoma -
.")
32
THANK YOU!

Еще похожие презентации в нашем архиве:







![Genetics Genetics (from Ancient Greek γενετικός genetikos, genitive and that from γένεσις genesis, origin),[1][2][3] a discipline of biology, is the.](/thumbs/17/1180906/big_thumb.jpg "Genetics Genetics (from Ancient Greek γενετικός genetikos, genitive and that from γένεσις genesis, origin),[1][2][3] a discipline of biology, is the.")









 distribution is a continuous probability distribution that has a bell-shaped probability.")

. Family How could you describe the word family? First of all family means a close unit of parents and their.")




 was a female domestic sheep, and the first mammal to be cloned from an adult.")
