Скачать презентацию
Идет загрузка презентации. Пожалуйста, подождите

1
ГЕНЕТИЧЕСКИЕ НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ ЧЕЛОВЕКА

2
Альбинизм- нарушение аминокислотного метаболизма. Врождённое отсутствие пигмента кожи, волос, радужной и пигментной оболочек глаза. Причиной альбинизма является отсутствие (или блокада) фермента тирозиназы, необходимой для нормального синтеза меланина особого вещества, от которого зависит окраска тканей. Они имеют повышенную чувствительность к солнечному свету, который вызывает у них воспалительные заболевания кожи.
 фермента тирозиназы, необходимой для нормального синтеза меланин")
4
Болезнь Кляйнфельтера- наличие лишней X хромосомы у мужчин (генотип – XXY). Для мужчин с синдромом Клайнфельтера характерны высокий рост, длинные конечности и относительно короткое туловище, евнухоидизм, бесплодие, гинекомастия, повышенное выделение женских половых гормонов, склонность к ожирению. Лишняя Х хромосома обусловливает различные нарушения психики. Больные очень внушаемы, вялы, апатичны, безынициативны, у них часто отмечается умственная отсталость (обычно дебильность). Нередко возникают параноидные, галлюцинаторно-параноидные, депрессивные психозы и навязчивые состояния, иногда наблюдаются антисоциальное поведение и алкоголизм. Клиническая картина начинает проявляться у мальчиков в период полового созревания. Диагностировать синдром Клайнфельтера, особенно у взрослых лиц, нетрудно, особенно при кариотипировании лишней Х хромосомы. Лечение проводится мужскими половыми гормонами для коррекции вторичных половых признаков.
. Для мужчин с синдромом Клайнфельтера характерны высокий рост, длинные конечности и относительно короткое туловище, евнухоидизм, бесплодие, гинекомастия, повышенное выделение")
6
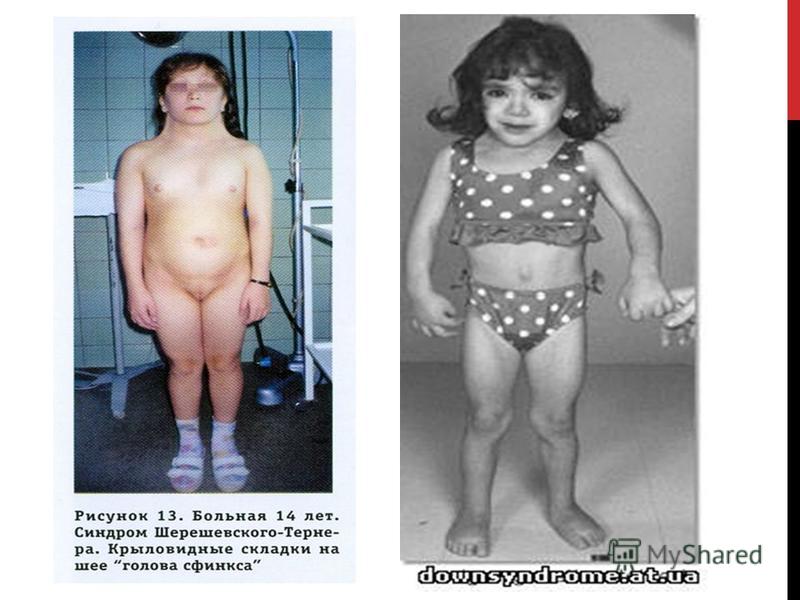
Синдром Шерешевского Тернера -сопровождающаяся характерными аномалиями физического развития, низкорослостью и половым инфантилизмом. Впервые эта болезнь как наследственная была описана в 1925 г. Н. А. Шерешевским, который считал, что она обусловлена недоразвитием половых желез и передней доли гипофиза и сочетается с врожденными пороками внутреннего развития. В 1938 г. Тернер выделил характерную для этого симптомокомплекса триаду симптомов: половой инфантилизм, кожные крыловидные складки на боковых поверхностях шеи и деформацию локтевых суставов. При синдроме Тернера половые железы обычно представляют собой недифференцированные соединительнотканные тяжи, не содержащие элементов гонад. Реже встречаются рудименты яичников и элементы яичек, а также рудименты семявыносящего протока. Другие патологические данные соответствуют особенностям клинических проявлений. Наиболее важны изменения костно-суставной системы укорочение пястных и плюсневых костей, аплазия (отсутствие) фаланг пальцев, деформация лучезапястного сустава, остеопороз позвонков. Рентгенологически при синдроме Тернера турецкое седло и кости свода черепа обычно не изменены. Отмечаются пороки сердца и крупных сосудов (коарктация аорты, незаращение боталлова протока, незаращение межжелудочковой перегородки, сужение устья аорты), пороки развития почек. Проявляются рецессивные гены дальтонизма и других заболеваний.

8
Синдром Кошачьего крика-Хромосомно синдром кошачьего крика объясняется частичной моносомией; он развивается при делеции (с утратой от трети до половины, реже полная утрата) короткого плеча пятой хромосомы. При этом синдроме наблюдается: общее отставание в развитии, низкая масса при рождении и мышечная гипотония, лунообразное лицо с широко расставленными глазами характерный плач ребёнка, напоминающий кошачье мяуканье, причиной которого является изменение гортани (сужение, мягкость хрящей, уменьшение надгортанника, необычная складчатость слизистой оболочки) или недоразвитие гортани. Признак исчезает к концу первого года жизни.
 короткого плеча пятой хромосомы. При этом синдроме наблюдается: общее отстава")
10
ПРОГЕРИЯ Большинство детей, больных прогерией, умирают в возрасте около 13-ти лет, но некоторые доживают и до 20- ти. Как правило, причиной смерти становится сердечный приступ или инсульт. В среднем, прогерия случается только у одного ребёнка из Заболевание вызвано мутациями в гене ламин A/C, белке, обеспечивающем поддержку клеточным ядрам. Другие симптомы прогерии включают жёсткую кожу, полностью лишённую волосяного покрова, костные аномалии, замедление роста и характерную форму носа. Прогерия представляет большой интерес для геронтологов, которые надеются выявить связь между генетическими факторами и процессом старения.

13
СИНДРОМ ЮНЕРА ТАНА (СЮТ) характерен прежде всего тем, что люди, страдающие им, ходят на четвереньках. Открыл его турецкий биолог Юнер Тан после изучения пяти членов семьи Улас в сельской местности Турции. Чаще всего люди с СЮТ пользуются примитивной речью и имеют врождённую мозговую недостаточность. В 2006-м году о семье Улас был снят документальный фильм под названием «Семья, ходящая на четвереньках». Тан описывает это так: «Генетическая природа синдрома предполагает обратную ступень в эволюции человека, вызванную, скорее всего, генетической мутацией, обратному процессу перехода от квадропедализма (хождения на четырёх конечностях) к бипедализму (хождению на двух). В этом случае синдром соответствует теории прерывистого равновесия. Новый синдром, по словам Тана, может быть использован в качестве живой модели человеческой эволюции. Некоторые исследователи, впрочем, не относятся к этому серьёзно и считают, что проявление СЮТ зависит не от генома.
 характерен прежде всего тем, что люди, страдающие им, ходят на четвереньках. Открыл его турецкий биолог Юнер Тан после изучения пяти членов семьи Улас в сельской местности Турции. Чаще всего люди с СЮТ пользуются примитивной")
14
ТЯЖЁЛЫЙ КОМБИНИРОВАННЫЙ ИММУНОДЕФИЦИТ Люди с этим генетическим отклонением рождаются без эффективной иммунной системы. Болезнь стала известна после вышедшего на экраны в 1976-м году фильма «Мальчик в пластиковом пузыре», вдохновлённого жизнью двух мальчиков- инвалидов Дэвида Веттера и Теда Де Виты. Главный герой, маленький мальчик, вынужден жить в изолированной от окружающего мира пластиковой кабинке, поскольку нефильтрованный воздух и воздействие микроорганизмов могут оказаться для него смертельными. Реальный Веттер смог прожить таким образом до 13-ти лет, но умер в 1984-м году после неудачной трансплантации костного мозга врачебной попытки укрепить иммунитет. Расстройство вызвано целым рядом генов, включая те, которые вызывают дефекты в Т и Б клеточных откликах, что в итоге оказывает негативное влияние на выработку лимфоцитов. Считается также, что это заболевание возникает в связи с отсутствием аденозиндезаминазы. Сейчас известны некоторые методы лечения с помощью генной терапии.

16
СИНДРОМ ПРОТЕЯ Вероятно, именно от этого заболевания страдал Джозеф Меррик, известный как Человек-слон. Синдром Протея вызван нейрофиброматозом типа I. При синдроме Протея кости и кожный покров больного могут начать увеличиваться аномально быстро, в результате чего нарушаются естественные пропорции тела. Обычно признаки заболевания не проявляются раньше 6–18 месяцев после рождения. Тяжесть заболевания зависит от индивидуума. В среднем синдромом Протея страдает один человек из миллиона. За всю историю задокументированной всего несколько сотен подобных случаев. Расстройство результат мутации в гене AKT1, ответственном за регуляцию клеточного роста, в результате чего некоторые мутировавшие клетки растут и делятся с невообразимой скоростью, а другие клетки продолжают расти в нормальном темпе. В итоге получается смесь нормальных и ненормальных клеток, что вызывает внешние аномалии. древнегреческой мифологии упоминается бог моря Протей, который имел свойство трансформировать форму своего тела. Именно его именем назвал данный синдром Ганс-Рудольф Видеман, немецкий педиатр.

18
ПОРФИРИЯ Порфирия это разновидность генетических патологий печени, при которых гемоглобин (красные кровяные тельца) синтезируется неправильно. В биосинтезе гемоглобина наличествует восемь ферментных шагов, и проблема с любым из них может явиться причиной порфирии. По словам доктора Дэвида Дольфина, известного специалиста по порфирии и другим болезням печени, люди, которых считали вампирами или оборотнями, могли страдать именно эти редким заболеванием. Дольфин утверждает, что на больного отрицательно влияет даже слабый солнечный свет. Повреждения кожи бывают такими серьезными, что нос или пальцы могут полностью разрушиться. Губы и десны могут значительно уменьшиться при сохранении нормальных размеров зубов в результате получается подобие звериной челюсти с клыками. К тому же у больных порфирией бывает усиленный рост волос. Дольфин пишет: …попробуйте представить, как в Средние века воспринимали того, кто выходил на улицу только по ночам, а вид его напоминал звериный повышенная волосатость, крупные зубы, обезображенное лицо. Предполагается (и это более чем вероятно), что таких людей вполне могли считать оборотнями. Дольфин предполагает, что вампиры-кровососы тоже были жертвами порфирии и «стремились ослабить симптомы своей страшной болезни». Если выпить много крови, то чужой гемоглобин принесет недостающие из-за нарушенного биосинтеза красные кровяные тельца и смягчит симптомы заболевания. Хотя эффект от гемоглобина, попадающего в кровь через стенки желудка, чрезвычайно мал. Сегодня больных порфирией часто лечат путем инъекций гемоглобина. В Средние века уколы были невозможны, поэтому потребление больших объемов крови являлось единственным способом, которым человек мог получить дополнительный гемоглобин. Больные порфирией отчаянно желали достать кровь, поскольку из-за нехватки гемоглобина наступала смерть. Неудивительно, что среди таких больных распространены патологические изменения личности и слабоумие.
 синтезируется неправильно. В биосинтезе гемоглобина наличествует восемь ферментных шагов, и проблема с любым из них может явиться прич")
20
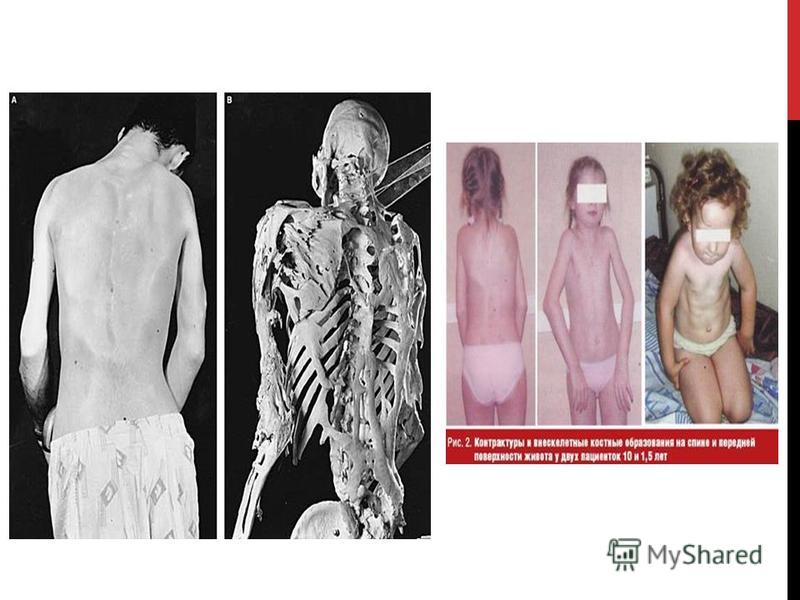
ФИБРОДИСПЛАЗИЯ Фибродисплазия оссифицирующая прогрессирующая или ФОП (от лат. fibro - волокно, dis - расстройство, нарушение, plasis - строение, структура и os, ossis - кость, facio - делать; окостенение) "мягкая соединительная ткань, которая прогрессивно превращается в кость. Очень редкое и тяжелое по своему течению генетическое заболевание, при котором мышцы, сухожилия и связки постепенно превращаются в кости. Процесс прогрессирует с годами, начинаясь обычно в пределах десятилетнего возраста у детей с мутацией определенного гена. Самые ранние зарегистрированные случаи относятся ко времени 17-х и 18-х столетий. В 1692 французский врач Гай Пэтин встретился с пациентом, который имел ФОП и упоминал встречу в своих письмах. В 1736, британец врач Джон Фрек описал подробно подростка, диагноз которого включал набухания всюду по его спине. Болезнь стала известной как миозит оссифицирующий прогрессирующий, что означает превращение мышцы прогрессивно в кость. Название было официально изменено на фибродисплазию оссифицирующую прогрессирующую в 1970-х доктором Виктором Маккьюзиком из Школы медицины Университета имени Джона Хопкинса, которого считают отцом современной медицинской генетики, в качестве обоснования он привел то что другие мягкие (или волокнистые - fibro) ткани в дополнение к мышцам (например, сухожилия и связки) могут быть затронуты окостенением. Также заболевание относится к врожденной наследственной патологии с аутосомно - доминантным типом наследования. Оно характеризуется неуклонно прогрессирующим течением, приводит к значительным нарушениям функционального состояния опорно-двигательного аппарата, глубокой инвалидизации больных и преждевременной их смерти, причем преимущественно в детском и молодом возрасте. Основу фибродисплазии составляет формирование воспалительных процессов в сухожилиях, связках, фасциях, апоневрозах и мышцах, что в конечном итоге приводит к их кальцификации и окостенению. Болезнь ещё называют «Болезнь второго скелета», так как по сути, там где в организме должны происходить штатные противовоспалительные процессы, начинается рост кости. У болезни нет расовой, половой, географической предрасположенности. Чаще всего болезнь возникает как результат спонтанной новой мутации.
")
22
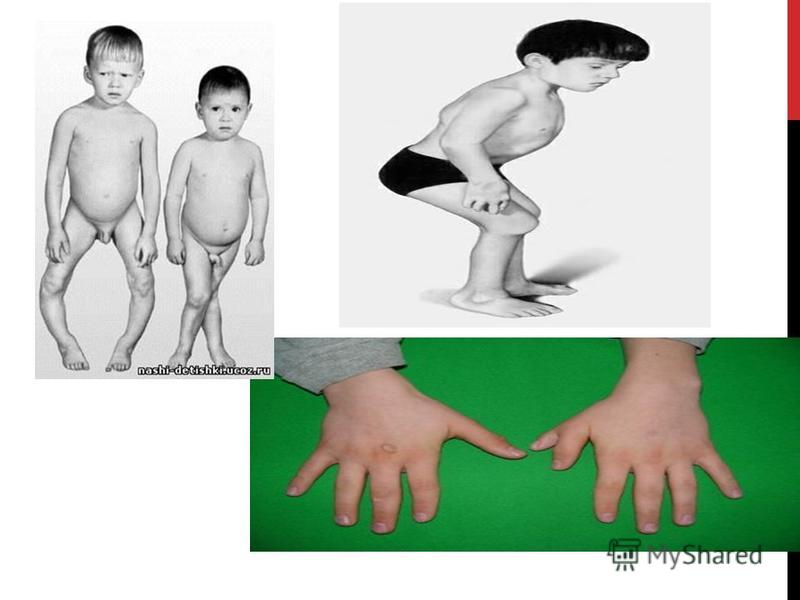
Рахит- дефект доминантного аллеля, вызывающий снижение содержания фосфора в крови. Короткопалость-Брахидактилия-аномалия развития по типу укорочения пальцев. Шестипалость-Полидактили́я-анатомическое отклонение, характеризующееся бо́льшим, чем в норме, количеством пальцев на руках или ногах у человека. Карликовость-Гипофизарный нанизм -это задержка роста и физического развития, вызванная недостаточным количеством соматотропного гормона (СТГ, гормон роста) в организме. Глухота-полное отсутствие слуха или такое его понижение, при котором невозможно разборчивое восприятие речи. Это потеря или резкое снижение слуха, при котором невозможно самостоятельное овладение устной речью.

Еще похожие презентации в нашем архиве:



















Мутационная 2)Комбинативная Цитоплазматическая.")
 Невозможность отбора и проведения направленного скрещивания. 2) Невозможность обеспечения одинаковых.")






 редкое наследственное заболевание нервной системы.")

