Скачать презентацию
Идет загрузка презентации. Пожалуйста, подождите

1
Общая характеристика моногенной патологии. Клиника и генетика отдельных форм моногенных болезней Проф. кафедры Павлишин Г.А.

2
Объемные образования потенциально возможные у новорожденного

3
Удельный вес наследственной патологии в детской смертности

4
Схема патогенеза наследственного нарушения обмена веществ (1) Ген АГен ВГен С Фермент АФермент ВФермент С Вещество А Вещество В Вещество С Вещество D
 Ген АГен ВГен С Фермент АФермент ВФермент С Вещество А Вещество В Вещество С Вещество D")
5
Ген А Ген В Ген С Фермент А Фермент В Фермент С отсутствует Вещество А Вещество В Вещество С Вещество X Вещество Y ВеществоZ Вещество D Схема патогенеза наследственного нарушения обмена веществ (2)
")
6
Клинические признаки врожденного нарушения обмена веществ и АК в постнатальном периоде Необъяснимое ничем отставание умственного и двигательного развития, судороги в анамнезе Необычный запах, особенно во время острого заболевания Интермитирующие эпизоды необъяснимой рвоты, ацидоза Гепатомегалия Почечные камни

8
Схема патогенеза фенилкетонурии (1) ФА Тирозин Меланин Адреналин Фенил-гидроксилаза
 ФА Тирозин Меланин Адреналин Фенил-гидроксилаза")
9
Схема патогенеза фенилкетонурии (2) ФА Накопление фенилаланина фенил-пировиноградной кислоты фенил-уксусной кислоты фенил-молочной кислоты Фенил-гидроксилаза
 ФА Накопление фенилаланина фенил-пировиноградной кислоты фенил-уксусной кислоты фенил-молочной кислоты Фенил-гидроксилаза")
10
Клинические признаки ФКУ Появление первых видимых признаков на 4-6 месяце Характерный внешний вид больного: бледная кожа, светлые волосы и глаза Неврологические нарушения: повышение сухожильных рефлексов, гипертонус мышц, симптом Грефе, гидроцефалия или микроцефалия Диспепсический синдром: частые рвоты, диарея Кожный синдром: бледная кожа, экзематозные проявления, повышенная потливость Специфический запах мочи и пота - мышиный, затхлый,плесени Изменения в психической сфере : пассивность, замедление реакций, речевые нарушения, агрессивное поведение

11
Фенилкетонурия у ребенка 1-го года. (значительная задержка психомоторного развития)
")
12
Подтвержающая (вторичная) диагностика ФКУ Метод тонкослойной и жидкостной хроматографии Молекулярно-генетические методы диагностики генного дефекта Определение наличия ФА и продуктов искаженного обмена в крови и моче ( проба Феллинга, индикаторные бумажные тесты) Полуколичественные микробиологические тесты ( тест Гатри, метод ауксотрофных штаммов E.Coli K-12)
 диагностика ФКУ Метод тонкослойной и жидкостной хроматографии Молекулярно-генетические методы диагностики генного дефекта Определение наличия ФА и продуктов искаженного обмена в крови и моче ( проба Феллинга, индикаторные бу")
13
Детские смеси,которые применяют при ФКУ

15
Целиакия ( глютеновая энтеропатия, идиопатическая стеаторея, нетропическая спру) Хроническое полисиндромное заболевание, которое характеризуется неспецифическими повреждениями слизистой оболочки тонкой кишки глютеном и проявляется нарушениями абсорбции на поврежденных участках кишечника
 Хроническое полисиндромное заболевание, которое характеризуется неспецифическими повреждениями слизистой оболочки тонкой кишки глютеном и проявляется нарушениями абсорбц")
16
Клинические критерии целиакии Развитие во втором полугодии жизни Наличие объемных, с неприятным запахом, светлых испражнений больше 2 раз в сутки Увеличение окружности живота, боли в животе Рвота ежедневная или периодическая Отставание в массе тела и роста Осалгии, повышенная ломкость костей Раздражительность, агрессивность, неспокойный сон Наличие жирных кислот, мыл в копрограмме Дисбактериоз кишечника

17
Лабораторные признаки целиакии Анемия Снижение уровня сывороточного железа и филатов Снижение содержания витамина В12 в сыворотке крови Гипоальбуминемия и гипергаммаглобулинемия Стеаторея Плоская кривая при проведении пробы на толерантность к глюкозе Рентгенконтрастное исследование тонкого кишечника - диффузное расширение кишечника, неровность складок слизистой оболочки Рентгенография скелета - признаки остеопороза

18
Схема патогенеза галактоземии (3) галактоза Накопление галактозо-1-фосфата -1-фосфат-уридилтрансфераза Галактозо Компенсаторное увеличение активности удфгп Второстепенный метаболизм галактозы Инактивация реабсорбции АК в почечных канальцах АК Вторичная аминоацидурия тестостерон
 галактоза Накопление галактозо-1-фосфата -1-фосфат-уридилтрансфераза Галактозо Компенсаторное увеличение активности удфгп Второстепенный метаболизм галактозы Инактивация реабсорбции АК в почечных канальцах АК Вторичн")
19
Диагностические критерии галактоземии Большая масса при рождении Диспепсические проявления сразу после рождения Быстрое развитие гипотрофии Желтуха с повышением неконъюгированного билирубина Гепатомегалия с дальнейшим развитием цирроза печени, портальной гипертензии Гипогликемия в анамнезе Отставание в психомоторном развитии Геморрагический диатез Формирование катаракты на третьей неделе жизни

20
Диагностика галактоземии Изучение генеалогического анамнеза Оценка анамнестических данных Выявление клинических диагностических критериев Определение гликемии с помощью различных методов Выявление галактозурии Снижение активности галактозо-1- фосфатуридилтрансферазы Изменение функциональных печеночных проб Оценка неврологического статуса Офтальмологическое исследование

21
Лечение галактоземии Исключение молока (галактозы) до 3 лет Назначение безлактозных смесей (Энфамил Соя, Нутрамиген, Симилак-изумил, Нутрисоя, Просоял) Прикорм вводят на месяц раньше, чем обычно с постепенной заменой смеси Медикаментозная терапия:1. Препараты, которые улучшают обмен галактозы 2. Урацил-4-карбоновая ( оротовая) кислота 3. Производные тестостерона 4. Препараты, стимулирующие ЦНС Сосудистые препараты Гепатопротекторы Антиоксиданты Витаминотерапия
 до 3 лет Назначение безлактозных смесей (Энфамил Соя, Нутрамиген, Симилак-изумил, Нутрисоя, Просоял) Прикорм вводят на месяц раньше, чем обычно с постепенной заменой смеси Медикаментозная терапия:1.")
22
Пероральноя нагрузка лактозой Введение перорально лактозы из расчета 1 г/кг массы тела ребенка Измерение содержания глюкозы в крови через 30 и 60 минут после нагрузки

23
Схема патогенеза І типа гликогеноза ( болезнь Гирке) гликоген Накопление гликогена в печени, почках -6-фосфатаза Глюкозо Вторичное нарушение липидного обмена Отложение жира в ПЖК Увеличение липидемии Увеличение печени и почек Гипогликемия
 гликоген Накопление гликогена в печени, почках -6-фосфатаза Глюкозо Вторичное нарушение липидного обмена Отложение жира в ПЖК Увеличение липидемии Увеличение печени и почек Гипогликемия")
24
Диагностические критерии болезни Гирке Клинические признаки гипогликемии (вялость, адинамия,рвота, судорожный синдром) Увеличение размеров живота Гепато-, спленомегалия, увеличение размеров почек Характерный внешний вид ребенка: круглое «кукольное» лицо, маленький рост, короткие руки и шея, значительно увеличенный в размерах живот, умеренное отложение жира в области лица и туловища, мышечная сила снижена Анорексия
 Увеличение размеров живота Гепато-, спленомегалия, увеличение размеров почек Характерный внешний вид ребенка: круглое «кукольное» л")
26
Диагностические критерии болезни Гирке Появление гипогликемии при недлительном голодании, натощак, поэтому больные должны постоянно принимать еду Гиперлипемия, гиперлактацидемия Проба с адреналином или глюкагоном отрицательная по гликемии Частая гиперкетонемия натощак с клиническими проявлениями кетозу, ацетонурия Нарушение функциональных проб печени Большое содержание гликогена в биоптатах печени и в клетках периферической крови, отсутствие, или снижения глюкозо-6-фосфотазы

27
Дагностические критерии болезнь Помпе Появление клиники в раннем грудном возрасте Шарообразная кардиомегалия Сердечная недостаточность по правожелудочковым типом Частые пневмонии на фоне ателектазов Характерный внешний вид ребенка: круглое, пастозное лицо, увеличенный язык, мышечная гипотония, задержка физического развития

29
ІІІ тип гликогеноза (гликогеноз печени и мышц, болезнь Форбса) обусловленный отсутствием фермента амино-1,6-глюкозидазы или олиго-1,1-глюкотрансферазы, что приводит к накоплению гликогена в печени, мышцах скелета и сердца
 обусловленный отсутствием фермента амино-1,6-глюкозидазы или олиго-1,1-глюкотрансферазы, что приводит к накоплению гликогена в печени, мышцах скелета и сердца")
30
Липидозы (lipidosis) Группа заболеваний, в основе которых лежит дефект определения фермента, который ведет к накоплению повышенного количества липидных субстанций в клетках и тканях ЦНС и внутренних органов тип наследования аутосомно-рецессивный
 Группа заболеваний, в основе которых лежит дефект определения фермента, который ведет к накоплению повышенного количества липидных субстанций в клетках и тканях ЦНС и внутренних органов тип наследования аутосомно-рецессивный")
31
Синдром Марфана Тип наследования аутосомно- доминантный Встречаются с частотой 1: Включает два типа: астенический (детский) и неастенический
 и неастенический")
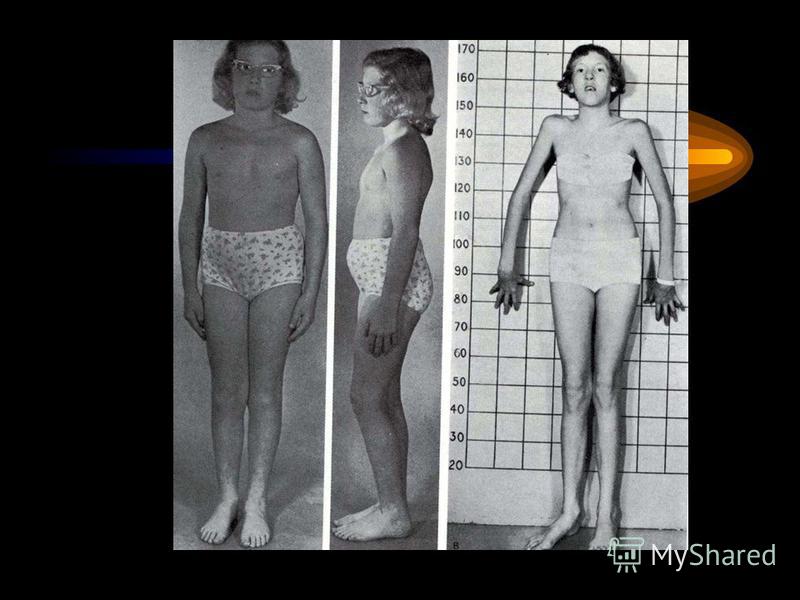
32
Два брата. Слева:нормальный мальчик 10 лет. Справа - 8-летний мальчик с синдромом Марфана (симптомы: подвывих хрусталика (он в очках), высокий рост, отсутствие подкожной жировой клетчатки, сколиоз, деформация грудной клетки)
, высокий рост, отсутствие подкожной жировой клетчатки, сколиоз, деформация грудной клетки)")
34
Диагностические критерии синдрома Марфана Нарушение мышечно-скелетной системы: арахнодактилия, высокий рост, длинные конечности, искривления позвоночника, деформация передней грудной клетки, ненормальная подвижность суставов, мышечная гипотония Кожный синдром: атрофичные стрии, паховые грыжи Патология глаз: миопия, вывих или подвывих хрусталика обеих глаз Сердечно-сосудистый синдром: расширение аорты, пролабирование митрального клапана, нарушение проведения возбуждения Бронхолегечной синдром: эмфизема, пневмоторакс

35
Ладони и ступни в норме (слева) и при синдроме Марфана (справа)
 и при синдроме Марфана (справа)")
36
Диагностические критерии арахнодактилии при синдроме Марфана

Еще похожие презентации в нашем архиве:



















 - это генетически обусловленная аномалия мезенхимального матрикса организма,")

 редкое наследственное заболевание нервной системы.")
