Скачать презентацию
Идет загрузка презентации. Пожалуйста, подождите

2
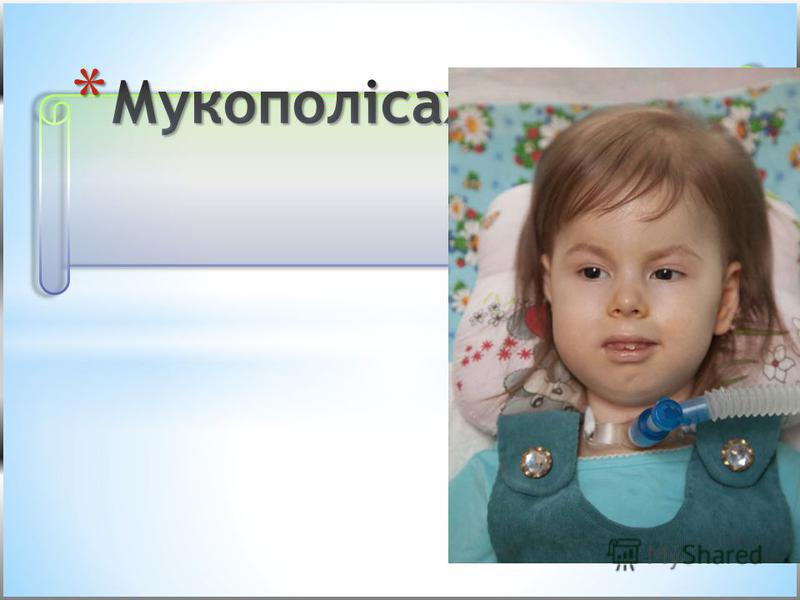
Це група спадкових хвороб сполучної тканини, обумовлена порушенням глікозаміногліканів (кислих мукополісахаридів) в результаті генетично обумовленої неповноцінності ферментів, що беруть участь в їх розчепленні. Успадковується за аутосомно-рецесивним типом.
 в результаті генетично обумовленої неповноцінності ферментів, що беруть участь в їх розчепленні. Успадковується за аутосомно-рецесивним")
3
* класифікація I тип синдром Хурлер, підтип --- синдром Хурлер- Шайя Обусловлені дефіцитом альфа-L-ідуронідази (фермент катаболізму мукополісахаридозів. Захварювання поступово призводить до накопичення в тканинах гепаринсульфату і дерматансульфату. Виділяють три фенотипи: синдром Хурлер, синдром Шая и синдром Хурлер-Шая.синдром Хурлерпідтип --- синдром Хурлер- Шайя II тип синдром Хантерасиндром Хантера III тип синдром Санфіліпосиндром Санфіліпо IV тип синдром Моркіосиндром Моркіо V тип синдром Шая VI тип синдром МаротоЛамі VII тип синдром Слая (дефицит β-глюкуронідази)синдром Слая VІІІ тип синдром Ди-Феран

4
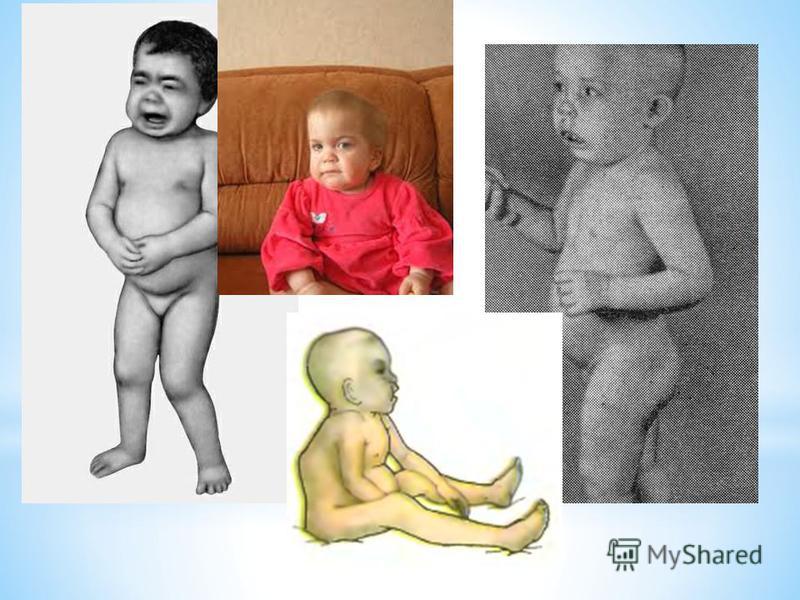
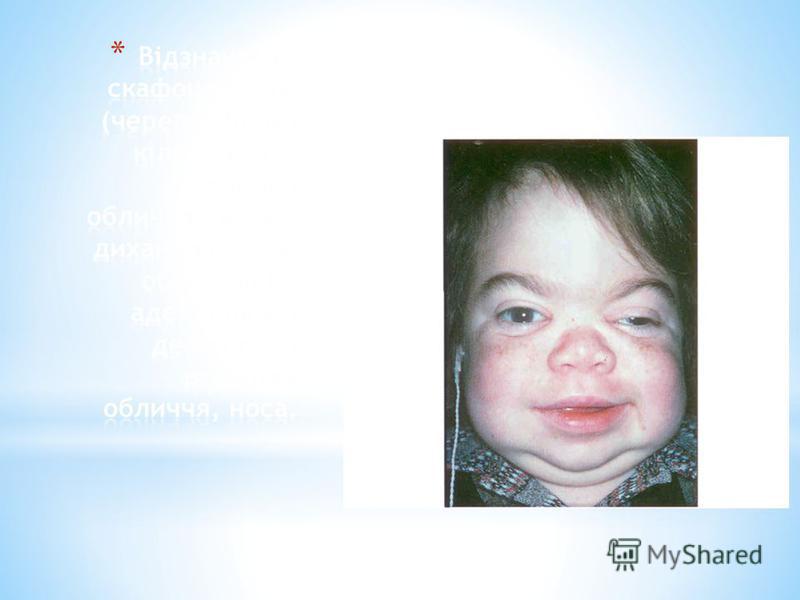
* Мукополісахаридоз типу I-Н (синдром Хурлер). * Вперше описаний німецьким педіатром G. Hurler в 1919 р. Часто спостерігається у дітей, батьки яких знаходяться в кровноспоріднених шлюбах.. * Проявляється на першому році життя, до 12 року клінічні прояви стають яскраво вираженими. * Поступово прогресує відставання в рості, формування деформованого скелету вкорочення шиї, випинання нижніх ребер, кіфоз грудного, поясничного відділу,, лопатки розміщені високо, кисті широкі, V палец короткий, викривлений. * Формуються згинальні контрактури починаючи з ліктьових та плечових суглобів, пізніше приєднуюьтся суглоби нижніх кінцівок, через це хворі ходять на напівзігнених ногах навшпиньки.. * Зміни зі сторони серця (систолічний шум,приглушені тони, розширення меж серця). Спостерыгаэться зниження слуху. * З віком наростає розумова відсталістьаж до стану, що нагадує ювенільну амавротичну ідіотію. * Спостерігається невротична симптоматика: підвищення тонусу мязів, паралічі порушення координації рухів.
. * Вперше описаний німецьким педіатром G. Hurler в 1919 р. Часто спостерігається у дітей, батьки яких знаходяться в кровноспоріднених шлюбах.. * Проявляється на першому році життя, до 12 року клінічні про")
11
* Вперше описаний американським офтальмологом Шайя (Н.G. Scheie) в 1962 р. * При народженні ознаки хвороби відсутні. Перші симптоми проявляються з 3-6 років Поступово розвивається обмеження рухів в суглобах. Обмеження рухів нихніх кінцівок незначні, можлива їх вальгусна деформація.
 в 1962 р. * При народженні ознаки хвороби відсутні. Перші симптоми проявляються з 3-6 років Поступово розвивається обмеження рухів в суглобах. Обмеження рухів нихніх кінцівок незначні,")
13
Мукополисахаридоз типа II (синдром Гунтера).
.")
15
Описаний американським педіатром Санфіліпо (S.J. Sanfilippo) в 1963 р. Частота 1 на новонароджених. Після народження впродовж 3-5 років дитина розвивається нормально, проте в деяких випадках спостерігаються незграбна хода, ускладнене ковтання. Перші симптоми хвороби у вигляді порушень сну з'являються у дітей старше 3 роки. Поступово розвивається апатія, знижується інтерес до іграшок, відзначається затримка психомоторного розвитку, порушення мови, риси обличчя грубіють. З'являються нетримання сечі і калу, діти перестають упізнавати оточення. Відзначаються також затримка росту, контрактури суглобів, гіпертрихоз, помірна гепатоспленомегалія. При рентгенологічному дослідженні кісткові зміни такі ж, як при синдромі Гурлер (але виражені слабко), або відсутні. На відміну від описаних вище типів при хворобі Санфіліпо в клінічній картині переважає розумова відсталість; ураження рогівки і серцево-судинної системи відсутні. Летальний кінець настає зазвичай в віці років через інфекційні чинники.
 в 1963 р. Частота 1 на 100 000-200 000 новонароджених. Після народження впродовж 3-5 років дитина розвивається нормально, проте в деяких випадках спостерігаються незграбна хода, ускладнене")
16
Захворювання в 1929 р. незалежно один від одного уперше описали уругвайський педіатр Моркіо (L. Morquio) і англійський радіолог Брейлсфорд (J.F. Braiisford). Частота до 1: Діти народжуються без ознак хвороби. Перші симптоми з'являються у віці 1-3 року, і до 7-8 років клінічна картина вже повністю виражена.
 і англійський радіолог Брейлсфорд (J.F. Braiisford). Частота до 1: 40 000. Діти народжуються без ознак хвороби. Перші симптоми з'являються у віці")
17
* Хворі карликового росту. Характерна особдивість: широкий рот, короткий ніс, широко поставлені зуби. Руки потворної форми, але суглоби досить рухливі. Шия коротка. Грудна клітка бочкоподібна, із стернальним кіфозом. З віком відбувається дифузне помутніння рогівки. Інтелект не міняється або помірно понижений. У деяких хворих в пізніх стадіях хвороби відзначається спастина параплегія і параліч дихальних м'язів. З сечею виділяється кератосульфат. Мукополісахаридоз типу IV (синдром Моркіо, хвороба Моркіо).

18
у 1960 р. уперше описаний французькими лікарями Марото (Р. Maroteaux) і Ламі (М. Е.J. Lamy). Перші симптоми з'являються у дітей старше за 2 роки.
 і Ламі (М. Е.J. Lamy). Перші симптоми з'являються у дітей старше за 2 роки.")
19
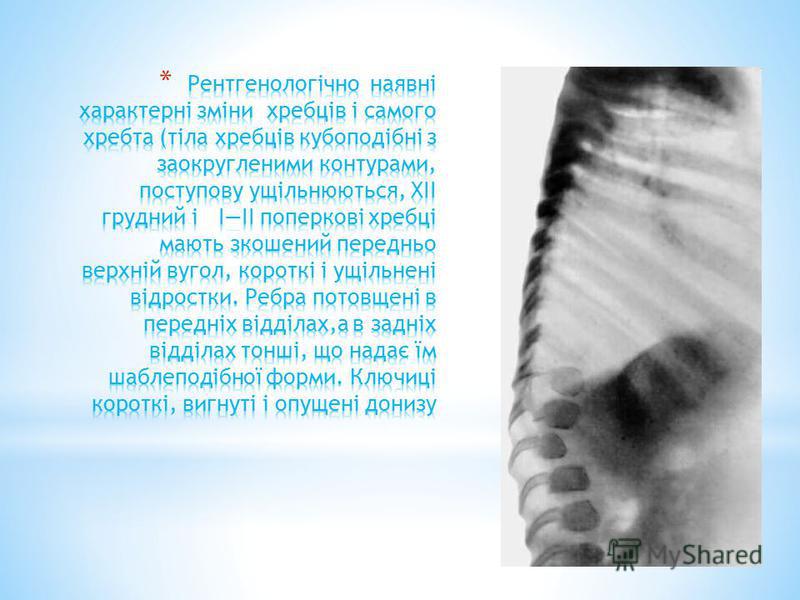
* При рентгенологічному дослідженні виявляються зміни хребців : в грудному - сколіоз, поперековому - кіфоз; у усіх відділах відзначається платиспондилия - сплощення і розширення тіл хребців, чим пояснюється характерне укорочення тулуба і незвично коротка шия. Змінюються кістки тазу : вертлужные западини плоскі і широкі, їх дах скошений, крила клубових кісток неправильної форми; контури усіх кісток нерівні.

20
* Характерне відставання в рості, грубі риси обличчя (як при синдромі Гурлер, але менш виражені), малі розміри верхньої щелепи, коротка шия, бочкоподібна грудна клітка, укорочені ключиці.
, малі розміри верхньої щелепи, коротка шия, бочкоподібна грудна клітка, укорочені ключиці.")
21
* Відзначаються згинальні контрактури суглобів верхніх кінцівок (хворі не можуть підняти руки вгору); з віком з'являються контрактури в суглобах нижніх кінцівок, порушується хода. Часто приєднуються гострі респіраторні вірусні інфекції. Нерідко виявляються грижі, гепатоспленомегалія. Можуть спостерігатися гідроцефалія, спастичні паралічі. Інтелект не страждає.
; з віком з'являються контрактури в суглобах нижніх кінцівок, порушується хода. Часто приєднуються гострі респіраторні вірусні інфекції. Нерідко виявл")
22
* Характеризується послабленням зв'язкового апарату суглобів, значними деформаціями скелета, зокрема грудним горбом. Спостерігаються також серцево- судинна недостатність, деформація нігтів, кривошия. Очні симптоми: дистрофічні зміни, помутніння строми рогівки, атрофія зорового нерва, Синдром Шая. Мукополісахаридоз типу - V.

23
Мукополісахаридоз типу VII (синдром Слая). Мукополісахаридоз типу VII (синдром Слая). Описаний Слаєм (W.S. Sly) в 1973 р. Клінічні прояви схожі з синдромом Санфіліпо. Діагноз встановлюють тільки при детальному біохімічному дослідженні. Мукополісахаридоз типа VIII (синдром Ді Ферранте). Мукополісахаридоз типа VIII (синдром Ді Ферранте). Описаний Ді Ферранте (N. Di Ferrante) та ін. в 1978 р. По клінічних проявах схожий з мукополисахаридозом типу IV (синдром Моркио), але на відміну від нього при мукополисахаридозе типу VIII виражена затримка психомоторного і інтелектуального розвитку.
. Мукополісахаридоз типу VII (синдром Слая). Описаний Слаєм (W.S. Sly) в 1973 р. Клінічні прояви схожі з синдромом Санфіліпо. Діагноз встановлюють тільки при детальному біохімічному дослідженні. Мукополісахари")
24
* Лікування та прогноз * Лікування симптоматичне. При цьому хворих спостерігають різні фахівці - хірурги (видалення гриж), ортопеди (ортопедична корекція порушень опорно-рухового апарату), педіатри (у зв'язку з частими гострими респіраторними вірусними інфекціями, серцево-судинною недостатністю), оториноларингологи (у зв'язку з порушеннями слуху, хронічними отитами і синуситами), офтальмологи, нейрохірурги і невропатологи (внутрішньочерепна гіпертензія). Використання для лікування гормональних препаратів (кортикотропіну, глюкокортикоїдів, тиреоїдину), вітаміну А, переливань препаратів крові плазми, введення декстрана- 70 приводить лише до тимчасового поліпшення. * Прогноз при усіх формах несприятливий, оскільки з віком наростають зміни скелета, порушення функцій різних органів і систем.
, ортопеди (ортопедична корекція порушень опорно-рухового апарату), педіатри (у зв'язку з частими гострими респіраторними вірусни")
Еще похожие презентации в нашем архиве:







. Частота 1:5000 1:7000 новонароджених. Клінічні діагностичні ознаки: -щілини верхньої.")







 СТЕФАНЮК ХРИСТИНА Синдром Патау або трисомія за 13 хромосомою.")
 тканин та органів людини, які здебільшого.")



 * Рухова (Виконують кістки кінцівок та хребта) * Підтримує певне вертикальне.")




